Columbia University Irving Medical Center
Transforming human health by driving discovery, advancing care and educating leaders

Our Schools
Advancing Care
News
- April 9, 2024
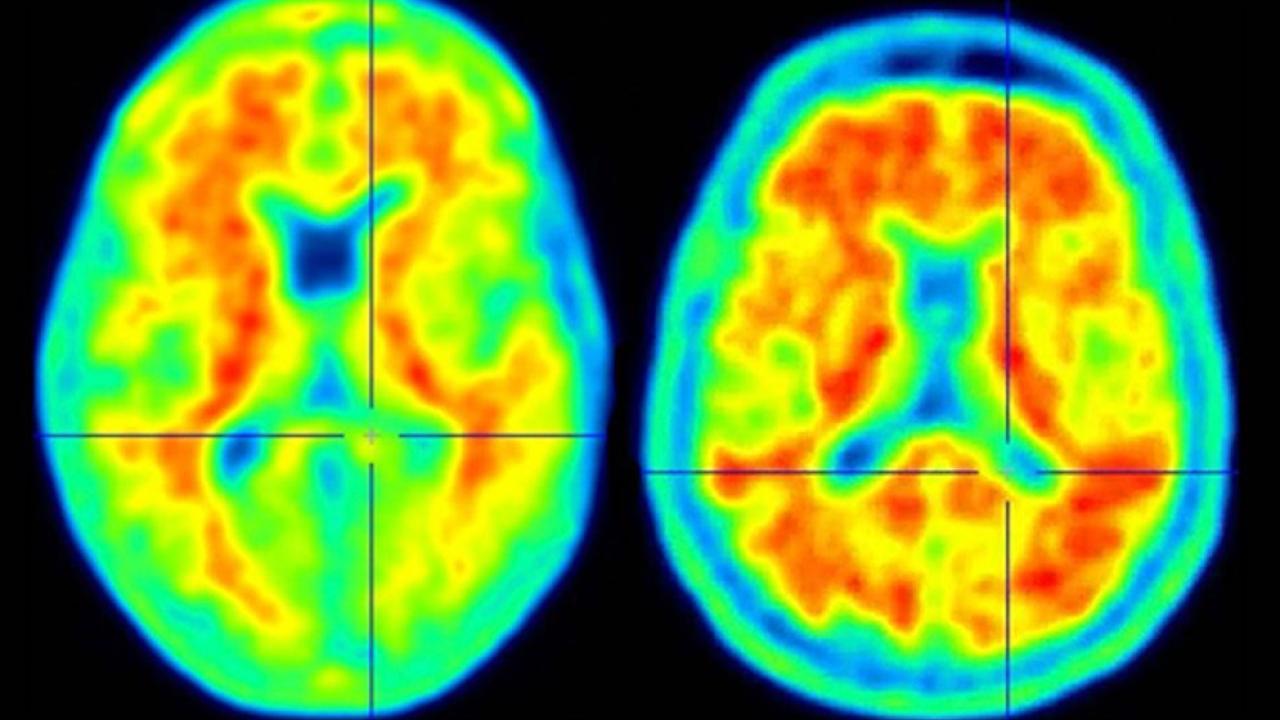
Columbia neuroscientists have identified a genetic mutation that fends off Alzheimer's in people at high risk and could lead to a new way to protect people from the disease.
Topic
- April 8, 2024
A new type of investigational therapeutic for pancreatic cancer has shown unprecedented tumor-fighting abilities in preclinical models of the disease.
Topic
- April 3, 2024
Columbia will begin construction in May on New York City’s first all-electric university research building.
Topic
- April 2, 2024
With funding from the Ultra-rare Gene-based Therapy Network, scientists at Columbia and the n-Lorem Foundation will create tailor-made gene-based therapies for people with rare forms of ALS.
Topic
- April 10, 2024
Young mothers have signs of faster cellular aging than other young women or young fathers, suggesting that pregnancy accelerates biological aging.
Topic




Our Community

Learn more about our role in serving the Northern Manhattan community of Washington Heights, Inwood, and Harlem.
Diversity, Equity, and Inclusion

At CUIMC, we are committed to providing culturally inclusive medical education, research, and clinical care.
Events
- Tuesday, April 16, 20249:00 AM to 10:00 AM
Venue
Online Event - Wednesday, April 17, 202412:00 PM to 1:00 PM
- Wednesday, April 17, 202412:00 PM to 2:00 PM
Venue
Haven Plaza, Haven Avenue between Fort Washington Avenue and 169th Street - Wednesday, April 17, 20246:00 PM to 7:00 PM
Venue
Online Event Online